Date: Jul 29, 2019, 14:00
Candidate: Nicola Leone da Rocha
Advisor: Rogério Custodio
Abstract:
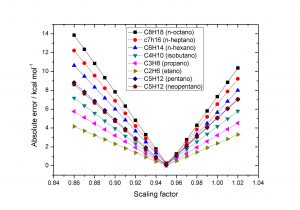
Composite methods are well-known computational simple methodologies capable of providing accurate results from corrections obtained at lower level calculations. Gaussian n theories or simply Gn belong to this class of methods and the energy of atoms and molecules are calculated from the general equation: E_{final}= E_{ref.} + ∆E_{Correlation} + ∆E_{Basis} + ∆E_{Difuse} + ∆E_{Polarization} +E_{HLC} + E_{SO} These steps are calculated using previously optimized geometries. Mean absolute errors of approximately 1.0 kcal/mol with respect to experimental data is one of the reasons for the popularity of these methods. These errors are obtained for properties such as enthalpies of formation, atomization energies, electron affinities, ionization energies, and proton affinities. The objective of this study was to analyze the influence of vibrational frequencies and geometry optimization on the enthalpies of formation. These evaluations were carried out by changing the levels of theory, basis functions, and the use of selective vibrational scaling factors in the frequency calculations. Results showed that the use of large basis functions can reduce the errors by up to 40% for hydrocarbons and substituted hydrocarbon. Other improvements were observed using different levels of theory, but not so significant. Our analysis on the vibrational scaling factors indicated that new perspectives should be considered and the treatment from the literature may not necessarily yield the best results. The procedure discussed in the present work showed that these factors are linear with respect to the enthalpy of formation errors, providing the determination of null errors when an optimal scaling factor is employed.