Guilherme F. Bottino
Institute of Chemistry and Center for Computing in Engineering and Sciences – University of Campinas, Brazil
![]()
Can we predict the functional shape of proteins? This is considered one of the most relevant challenges in computational biology, and was selected as one of the major problems of science in the 21st century by Science magazine in its 125 year commemorative edition (Kennedy & Norman, Science 1, 309, 2005). This challenging task requires the execution of complicated calculations on powerful supercomputers, but this is far from being its main limitation: in most cases the available structural data is not enough to determine their fold.
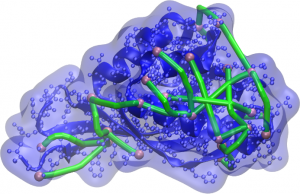
Representation of the protein and its surface (blue) with molecular rulers (green) connecting surface-accessible atoms (pink). Computer-generated models should ensure that the distances between these atoms are not greater the linker length.
Addressing such problem, researchers from the CCES (University of Campinas, Brazil), decided to improve protein structure modeling methods by aggregating experimental information to their computational calculations. Aided by collaborators within the university itself, like Prof. Fabio Gozzo (Dalton MS Group), experiments employing molecular rulers were carried out, which allow for the estimation of distances between different atoms on the protein surface. Those distances were translated as constraints to the maximum separation of those atoms, and provided to the modeling algorithms, leading to improvement of success rates or reduction of the time required to propose a three-dimensional protein structure. The combination of several restrictions amplifies the beneficial effects.
To understand this phenomenon, think that molecular rulers – also called linkers – receive this name because they are molecules of known length with reactive ends, capable of chemically combining to side chains of amino acids in specific ways. Proteins, in turn, put on a specific shape in their natural state, allowing some atoms on their surface to be exposed, accessible to small molecules around them. By mixing molecular rulers and proteins, a connection can occur between two atoms if they are within the well-defined range of a linker. By means of a technique called Cross-linking Mass Spectrometry (XLMS), it is possible to detect connections and which are the connected atoms; if the extended size of the linker is known, scientists can propose that the generated 3D models should be consistent with this maximum separation distance.
“For several years, protein structure determination guided only by this type of information has been a great challenge,” says Professor Leandro Martínez. “We have developed new ways of describing the linkers in computers, improved validation of experimental data and three-dimensional models, and proposed methods for data selection and interpretation. In some cases, the quality increase in protein modeling outputs was 400 times superior, if compared to the classical unconstrained modeling approach”. Prof. Leandro is one of the principal investigators in the biomolecular modeling line of research at the CCES, funded by Fapesp.
One of these improvement points was described in an article published in January 2019, “Statistical force-field for structural modeling using chemical cross-linking / mass spectrometry distance constraints”. It brought innovation to the way constraints are understood and incorporated into the computational protocol of model generation. Statistical techniques allowed the suggestion of new mathematical expressions capable of describing constraints more precisely, greatly increasing the quality of output models. Such result not only represents further advance in the universal effort to improve protein modeling techniques, but also endorses the power of bilateral collaboration between theoretical and experimental science, mutually filling in their gaps.
Associated Scientific Research:
A. J. R. Ferrari, F. C. Gozzo, L. Martínez, Statistical force-field for structural modeling using chemical cross-linking/mass-spectrometry distance constraints.
Bioinformatics, 2019.[Full Text] [XLFF site]
A. J. R. Ferrari, M. A. Clasen, L. Kurt, P. C. Carvalho, F. C. Gozzo, L. Martínez, TopoLink: Evaluation of structural models using chemical crosslinking distance constraints. Bioinformatics, 2019. [Full Text] [TopoLink site]
R. N. Santos, X. Jiang, L. Martínez, F. Morcos, Coevolutionary Signals and Structure-Based Models for the Prediction of Protein Native
Conformations. In: Computational Methods in Protein Evolution, Methods in Molecular Biology. vol. 1851. Springer 2019.[LINK]
L. Censoni, L. Martínez, Prediction of kinetics of protein folding with non-redundant contact information.
Bioinformatics, 34(23), 4034-4038, 2018. [LINK] [Associated software]