Guilherme F. Bottino
Instituto de Química e Centro de Computação em Engenharia e Ciências – Universidade de Campinas
![]()
Podemos prever o formato funcional das proteínas? Essa é considerada uma das questões mais relevantes da biologia computacional, e foi selecionada como um dos grandes problemas da ciência do século XXI pela revista Science em sua edição comemorativa de 125 anos (Kennedy & Norman, Science 1, 309, 2005). A desafiadora tarefa exige a execução de cálculos complicados em poderosos supercomputadores, mas essa está longe der ser sua principal limitação: na maior parte dos casos os dados conhecidos sobre a estrutura não são suficientes para sua determinação.
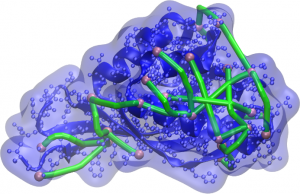
Representação da proteína e sua superfície (em azul) com as réguas moleculares (em verde) conectando átomos acessíveis (em rosa). Modelos gerados pelo computador devem garantir que as distâncias entre esses átomos não sejam maiores que a permitida pelo conector.
Pensando nisso, pesquisadores do CCES, da Universidade Estadual de Campinas (Unicamp), decidiram aprimorar os métodos de modelagem da estrutura de proteínas pela incorporação de informações experimentais aos cálculos computacionais. Com ajuda de colaboradores da própria universidade, como o Prof. Fabio Gozzo (Dalton MS Group), foram realizados experimentos com réguas moleculares que permitem estimar as distâncias entre diferentes átomos na superfície da proteína. Essas distâncias são traduzidas como restrições à separação máxima desses átomos e fornecidas aos algoritmos de modelagem, levando ao aumento das taxas de sucesso ou à diminuição do tempo necessário para proposição de uma estrutura tri-dimensional para as proteínas. A combinação de várias restrições amplifica os efeitos benéficos.
Para entender esse fenômeno, basta pensar que réguas moleculares – também chamadas de conectores – recebem esse nome por serem moléculas com comprimento conhecido e duas extremidades reativas, capazes de se combinar quimicamente às cadeias laterais dos aminoácidos de forma específica. As proteínas, por sua vez, se organizam num formato específico em seu estado natural, permitindo que alguns átomos na sua superfície fiquem expostos, acessíveis a moléculas pequenas ao seu redor. Ao misturar réguas moleculares e proteínas, pode ocorrer uma conexão entre dois átomos se eles estiverem dentro do alcance bem definido de um conector. Por meio de uma técnica chamada Espectrometria de Massas de Cross-linking, é possível determinar se há conexões e quais são os átomos conectados, e sabendo o tamanho da régua molecular empregada, pode-se propor que os modelos gerados sejam consistentes com essa distância máxima de separação.
“Há vários anos, a modelagem de estruturas de proteínas guiada apenas por esse tipo de informação é um grande desafio”, diz o professor Leandro Martínez. “Nós abordamos diferentes problemas associados ao uso dessas restrições para otimizar o processo como um todo. Desenvolvemos novas formas de descrever as réguas moleculares nos computadores, melhoramos a validação dos dados experimentais e dos modelos tridimensionais gerados, e propusemos métodos de interpretação e seleção de dados. Em alguns casos, o incremento na qualidade das modelagens de proteína foi superior a 400 vezes, em relação ao processo clássico de modelagem sem restrições.” O Prof. Leandro é um dos pesquisadores principais da linha de pesquisa em modelagem molecular no Centro de Computação em Engenharia e Ciências (CCES), financiado pela Fapesp.
Um desses pontos foi explorado em um artigo científico publicado em janeiro de 2019, “Statistical force-field for structural modeling using chemical cross-linking/mass spectrometry distance constraints”. Trata-se de uma inovação na maneira como as restrições são entendidas e incorporadas ao protocolo computacional de geração dos modelos. Técnicas estatísticas permitiram sugerir novas expressões matemáticas capazes de descrever as restrições de distância mais precisamente, aumentando muito a qualidade dos modelos obtidos. Esse tipo de resultado é importante não só por representar mais um avanço no esforço universal de melhoria das técnicas de modelagem de proteínas, mas também por exemplificar o poder da colaboração bilateral e de qualidade entre ciência teórica e experimental, preenchendo mutuamente suas lacunas.
Artigos científicos:
A. J. R. Ferrari, F. C. Gozzo, L. Martínez, Statistical force-field for structural modeling using chemical cross-linking/mass-spectrometry distance constraints.
Bioinformatics, 2019.[Full Text] [XLFF site]
A. J. R. Ferrari, M. A. Clasen, L. Kurt, P. C. Carvalho, F. C. Gozzo, L. Martínez, TopoLink: Evaluation of structural models using chemical crosslinking distance constraints. Bioinformatics, 2019. [Full Text] [TopoLink site]
R. N. Santos, X. Jiang, L. Martínez, F. Morcos, Coevolutionary Signals and Structure-Based Models for the Prediction of Protein Native
Conformations. In: Computational Methods in Protein Evolution, Methods in Molecular Biology. vol. 1851. Springer 2019.[LINK]
L. Censoni, L. Martínez, Prediction of kinetics of protein folding with non-redundant contact information.
Bioinformatics, 34(23), 4034-4038, 2018. [LINK] [Associated software]